|
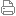
自然界生物体中广泛存在一类活性中心含有两个铁原子的双铁加氧酶。研究双铁加氧酶含氧活性中间体的结构,对揭示其活化O2的机制非常关键。然而,由于含氧活性中间体的活性高/不稳定,用传统的X-ray单晶衍射方法获取其结构非常困难,通过理论化学预测双铁加氧酶含氧活性中间体的结构,成为当前该领域的重要研究前沿。 在中科院化学所理论计算化学平台项目和国家自然科学基金委的支持下,化学所光化学院重点实验室的陈辉等在理论计算预测双铁加氧酶含氧活性中间体结构研究方面取得系列进展。在前期工作中,针对蓝藻醛去甲酰化加氧酶(cADO),科研人员对其含氧活性中间体结构、57Fe穆斯堡尔谱参数、底物氧化机理等,开展了多尺度QM/MM(混合量子力学/分子力学)理论计算模拟研究,证实了其中间体结构是O2与醛底物形成的过氧半缩醛结构(图1),而不是以往通常认为的过氧化物结构。研究还发现,O2、醛底物、对称双铁中心三者间的相互作用,具有左右不对称性(图1),在含氧活性中间体形成时,作为双铁配体的一个组氨酸残基会罕见地从双铁中心解离。该工作也显示QM/MM水平的57Fe穆斯堡尔谱模拟,在双铁加氧酶含氧活性中间体预测方面具有应用潜力。相关结果发表在J. Phys. Chem. Lett. 2016, 7, 4427-4432。 图1 计算得到的cADO含氧活性中间体结构及其57Fe穆斯堡尔谱参数 最近,针对目前仅有的两种双铁芳胺加氧酶AurF和CmlI,利用多尺度QM/MM模拟结合57Fe穆斯堡尔谱模拟,科研人员首次提出了这两种酶含氧活性中间体P具有类似的结构。研究发现:(1)AurF和CmlI的含氧活性中间体P都具有μ-η0:η2型O2配位的双三价铁氢过氧化物((FeIII)2-hydroperoxo)结构(图2);(2)预测的双三价铁氢过氧化物结构P可以有效介导芳胺基到亚硝基,再到硝基的芳胺氮氧化过程,其中涉及胺基孤对电子的电子耦合质子转移(PCET)过程对胺基N-H键断裂起重要作用;(3)理论计算预测的双三价铁氢过氧化物的μ-η0:η2型O2配位结构可以解释很多双铁芳胺加氧酶实验研究中的难题,如AurF和CmlI不同的57Fe穆斯堡尔谱参数,含氧活性中间体P相对较低的O-O振动频率、含氧活性中间体P的亲电/亲核双亲反应性、以及其C-H活化的反应惰性。科研人员预言的双铁芳胺加氧酶AurF和CmlI含氧活性中间体P的结构类似性,为统一它们的酶催化反应机理提供了基础。相关结果发表在J. Am. Chem. Soc. 2017, 139, 13038-13046。 图2 理论计算预测得到的双铁芳胺加氧酶AurF和CmlI的含氧活性中间体结构
| 







 发表于 2017-10-12 17:50:18
发表于 2017-10-12 17:50:18

 QQ好友和群
QQ好友和群 QQ空间
QQ空间 腾讯微博
腾讯微博 腾讯朋友
腾讯朋友 收藏
收藏 转播
转播 分享
分享 淘帖
淘帖