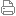
钯催化烯丙基取代反应是构建碳-碳和碳-杂原子键的有力工具,广泛应用于天然产物和药物的合成中。上海交通大学张万斌教授课题组在过去的几年中开发了一系列基于新型烯丙基底物和亲核试剂的烯丙基取代反应,取得了优良的催化效果(J. Am. Chem. Soc., 2011,133, 19354; Angew. Chem. Int. Ed.,2014, 53, 6776; J. Am. Chem. Soc., 2016,138,11093-11096; J. Am. Chem. Soc., 2017, 139, 9819; J. Am. Chem. Soc., 2018,140, 2080-2084)。令人欣喜的是,他们采用有机小分子或者金属与亲核试剂前体原位络合形成活化的中间体,再与烯丙基底物反应,成功构建了非天然氨基酸和多肽等活性分子。 哌啶杂环结构单元广泛存在于药物和天然产物中,其高效构建也一直是一个富有挑战性的课题。从便宜易得的吡啶芳环出发,经过活化后再官能化构建复杂哌啶结构一直是哌啶类化合物最重要的合成方法之一。最常见的活化吡啶芳环方法就是形成吡啶季铵盐,借助吡啶阳离子的吸电子效应使其化学性质更加活泼,因而可以应用于更为丰富的反应类型。吡啶季铵盐可以参与许多反应,例如迈克尔加成、1,3-偶极加成、Kröhnkereaction反应和还原反应等。吡啶季铵盐中的吡啶基团在反应过程中可以直接脱去,也可以被还原而衍生为哌啶类衍生物,是一种十分有价值的合成子。尽管人们对吡啶季铵盐的研究已经取得了许多进展,但是通过取代反应对吡啶季铵盐的α-位进行官能化至今还未见诸报道。在本文中,作者认为如果能将α-卤代羰基化合物和吡啶原位生成的吡啶季铵盐作为活化的亲核试剂应用于钯催化不对称烯丙基取代反应,再进一步将吡啶结构还原为哌啶,则可以得到一系列具有潜在生物活性的含哌啶结构单元的非天然氨基酸衍生物,进一步扩展哌啶类小分子的结构多样性。 张万斌团队首先采用了α-卤代酮和酯进行反应,发现尽管可以获得较高收率,却得到消旋的产物。他们认为这可能是由于酮和酯的α-位酸性较强,生成的产物易于消旋化造成的。因此,他们选用了α-位酸性较弱的酰胺作为底物,以课题组开发的面手性二茂铁配体作为手性配体进行试验,最终筛选出优化的反应条件:手性配体L1,碱为TMG,溶剂为混合的二氧六环与二甲亚砜(10/1,v/v),离去基团为碳酸甲酯OCO2Me,添加剂为碘化锂,反应温度为5 oC。 在上述优化的反应条件下,作者对烯丙基底物适用性进行了考察。发现苯环上无论是吸电子还是供电子基团,反应均以较高的收率和对映选择性获得目标产物。对苯环上不同位置取代基的考察表明,采用4-位取代底物获得的对映选择性优于2-位和3-位。当芳环被氢或单个烷基取代时,反应仍可以顺利进行,给出较好的收率和对映选择性。当底物上有双烷基取代基时,产物的ee值不理想。 张万斌进一步对吡啶杂环的底物适用性进行了考察。结果发现,无论吡啶环上4-位是烷基还是芳基官能团都能以良好的收率和对映选择性得到目标产物。当吡啶环上3-位有取代基时,反应也可以顺利反应,但是对映选择性略有降低。
| 







 发表于 2018-12-16 09:20:19
发表于 2018-12-16 09:20:19

 QQ好友和群
QQ好友和群 QQ空间
QQ空间 腾讯微博
腾讯微博 腾讯朋友
腾讯朋友 收藏
收藏 转播
转播 分享
分享 淘帖
淘帖